اساس ژنتیکی و تشخیص بالینی هایپر کلسترولمی خانوادگی
به گزارش روابط عمومی پایگاه اطلاع رسانی علوم آزمایشگاهی ایران، هایپرکلسترولمیای خانوادگی& یا FH یکی از شایع ترین و شناخته شده ترین اختلالات ژنتیکی است. جهش در ژن گیرنده لیپوپروتئین با چگالی کم& یا LDLR در بیش از 90٪ موارد علت اصلی بیماری است. FH با ریسک بالای بیماری عروق کرونر قلب زودرس یا PCAD همراه است. شیوع FH& از 1 در 200 تا 1 در 500 نفر جمعیت تخمین زده می شود. با این حال بر اساس برخی مطالعات، گزارش شده که بیش از 80٪ افراد مبتلا به FH تشخیص داده نشده باقی می مانند.
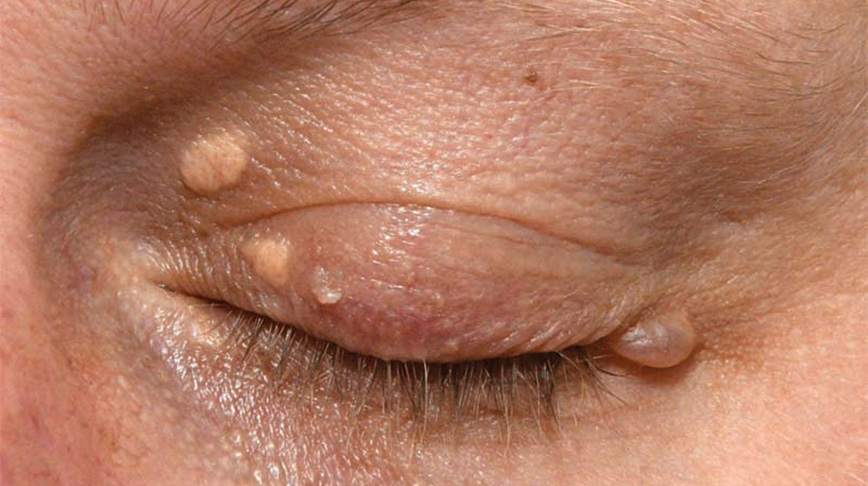
تظاهرات اصلی FH شامل افزایش قابل توجه سطح سرمی LDL-C، بیماری عروق کرونر زودرس& یا ASCVD و رسوب کلسترول در زیر پوست (زانتوما ) و قوس قرنیه می باشد. سطح LDL-C به طور معمول برای سن و جنس به بیش از صدک 90 افزایش می یابد، در حالی که سطح لیپوپروتئین با چگالی سنگین& یا HDL-C و تری گلیسیرید معمولاً طبیعی است یا به مقدار کمی تغییر می کند. دلایل ثانویه هایپر کلسترولمی، مانند اختلال عملکرد کبدی، کلیوی و تیروئید، باید کنار گذاشته شود. برخی از داروها ممکن است منجر به افزایش ثانویه سطح LDL-C شود.
پاتو فیزیولوژی اصلی FH به طور کلی بعلت جهش در ژن LDLR و به میزان کمتر در ژن های آپو لیپوپروتئین B (APOB) و PCSK9 رخ می دهد. ژن LDLR روی کروموزوم 19 قرار دارد و دارای 18 اگزون با طول 45 کیلوباز است. تاکنون بیش از 1288 جهش منحصر به فرد و واریانت های اللی در ژن LDLR شناسایی شده است. جهش در ژن هایی که پروتئین های درگیر در جذب LDL و کاتابولیسم را رمزگذاری می کنند، یعنی LDLR، APOB، پروتئین آداپتور گیرنده LDL یا LDLRAP1 و& PCSK9& شناخته شده اند که باعث ابتلا به FH می شوند، ناشی از برداشت و تنظیم معیوب LDL می باشند.
معیارهای بالینی مختلفی برای تشخیص FH ارائه شده است. در عمل، دو گروه از معیار های بالینی معتبر، به عنوان مثال، سیستم& DLCN و سیستم& Simon Broome (SBR) بر اساس سابقه بالینی خانوادگی و فردی، معاینه فیزیکی و نتایج پروفایل لیپیدی، به طور گسترده ای برای شناسایی افراد مبتلا استفاده می شود. در جمعیت های آسیایی، معیارهای فوق الذکر با تغییراتی مورد قبول قرار گرفته اند. این مجموعه معیارها ممکن است سبب عدم تشخیص برخی از بیماران هتروزیگوت مبتلا به نوع فنوتیپ خفیف گردد و برای جمعیت کودکان نیز قابل استفاده نیستند. با اینحال، هر دو معیار فوق الذکر در بالین برای تشخیص FH کاربرد دارند.
اکثر موارد FH به علت جهش در ژن های LDLR، APOB& و PCSK9 ایجاد می شود. جهش بیماری زا در یکی از این ژنها در حدود 70 درصد موارد قطعی FH& با فنوتیپ مشخص و 20 درصد موارد با تشخیص احتمالی FH شناسایی شده است. تکنیک های جدید مولکولی، مانند تعیین توالی اگزوم، می تواند منجر به کشف جهش های جدید شود. این تکنیک به ویژه در مورد جمعیت های چند قومی می تواند مورد مطالعه قرار گیرد. حدود 95 درصد جهش های شناسایی شده در ژن APOB و 1 درصد در ژن PCSK9 قرار دارند. تشخیص جهش در یکی از اعضای خانواده کمک می کند تا تشخیص قطعی FH انجام شود. با این حال، عدم تشخیص جهش، تشخیص FH را نفی نمی کند، به ویژه اگر فنوتیپ بالینی به احتمال زیاد بیانگر وجود FH باشد. خدمات غربالگری متمرکز ژنتیکی همراه با رهنمود هایی برای بازپرداخت بیمه ها میتواند میزان انجام آزمایش ژنتیکی و غربالگری آبشاری را بهبود بخشد. تلاش ها برای توسعه برنامه های آموزشی برای مشاوران ژنتیک و محققان لیپید در مورد ژنتیک FH ادامه دارد.
درمان هایپر کلسترولمی خانوادگی
در حال حاضر، استاتینها، ازتیمایب، گیرنده های اسید صفراوی& و مهار کننده های PCSK9 اصلی ترین روش های درمانی برای درمان هایپرکلسترولمی هستند. علاوه بر این، درمان های جدید مبتنی بر RNA نظیر Inclisiran تأثیر بسیار زیادی در استراتژی های درمانی دهه های اخیر داشته است. درمان باید به محض تشخیص آغاز شود و در طول زندگی ادامه یابد. در حالی که هایپرکلسترولمی ممکن است در اثر تغییرات خاص ژنتیکی ایجاد شود، بیشتر موارد هایپر کلسترولمی تحت تأثیر عوامل دیگری مانند بیماری های غدد درون ریز یا متابولیک، سبک زندگی (ورزش و رژیم) و داروها قرار دارند. این دارو ها از طریق افزایش فعالیت گیرنده LDL و پاکسازی LDL-C عمل می کنند. علاوه بر این، با پیشرفت چشمگیر تکنیک های مولکولی، رویکرد های درمانی مبتنی بر RNA برای مداخله درمانی تأثیر زیادی بر استراتژی های درمانی دهه های اخیر داشته است.